SAIGE

SAIGE
SAIGE(Scalable and Accurate Implementation of GEneralized mixed models)是一个用于大规模基因组关联研究(GWAS)的 R 包,旨在通过广义混合模型(Generalized Mixed Models, GMM)处理样本之间的相关性,并对大规模遗传数据进行高效分析。SAIGE 具有处理大量样本、考虑遗传关系、避免样本相关性干扰等优点,是解析全基因组数据的理想工具。
本文将介绍 SAIGE 的主要功能、使用场景及其工作原理。
1. SAIGE 的主要特点
1.1 高效的计算方法
SAIGE 的核心优势之一是其高效的计算方法。它能够处理大规模数据集,尤其适用于大约几十万到上百万的样本。SAIGE 通过采用广义混合模型(GMM)来估计样本之间的遗传相关性,从而准确估计每个基因位点的效应大小,并进行全基因组关联测试。
1.2 处理样本相关性
在 GWAS 中,样本之间的相关性(例如家族结构或人口结构)可能会导致结果的偏差。SAIGE 通过基因型关系矩阵(GRM)来处理样本的相关性,使得 GWAS 结果更加可靠。无论是亲缘关系还是其他类型的样本相关性,SAIGE 都能够有效地调整,避免不必要的假阳性结果。
1.3 支持多种表型类型
SAIGE 不仅支持定量性状(例如身高、体重等),还支持二元性状(例如病例对照数据)。它能够处理不同类型的表型,并针对不同表型类型应用相应的统计方法,例如,Firth 的偏差减少逻辑回归(Firth’s Bias-Reduced Logistic Regression)来分析稀有变异。
1.4 稀有变异分析的扩展功能(SAIGE-GENE+)
SAIGE-GENE+ 是 SAIGE 的扩展,用于稀有变异的集成分析。它提供了多种常用的稀有变异测试方法,包括 BURDEN 测试、SKAT 测试和 SKAT-O 测试,帮助识别变异集与表型的关系。
1.5 高度可定制化
SAIGE 允许用户根据实际需求调整参数,例如显著性阈值、遗传关系矩阵类型(完整或稀疏)等。此外,它还支持条件分析,可以在现有 GWAS 信号的基础上进行进一步的变异筛选。
2. SAIGE 的工作原理
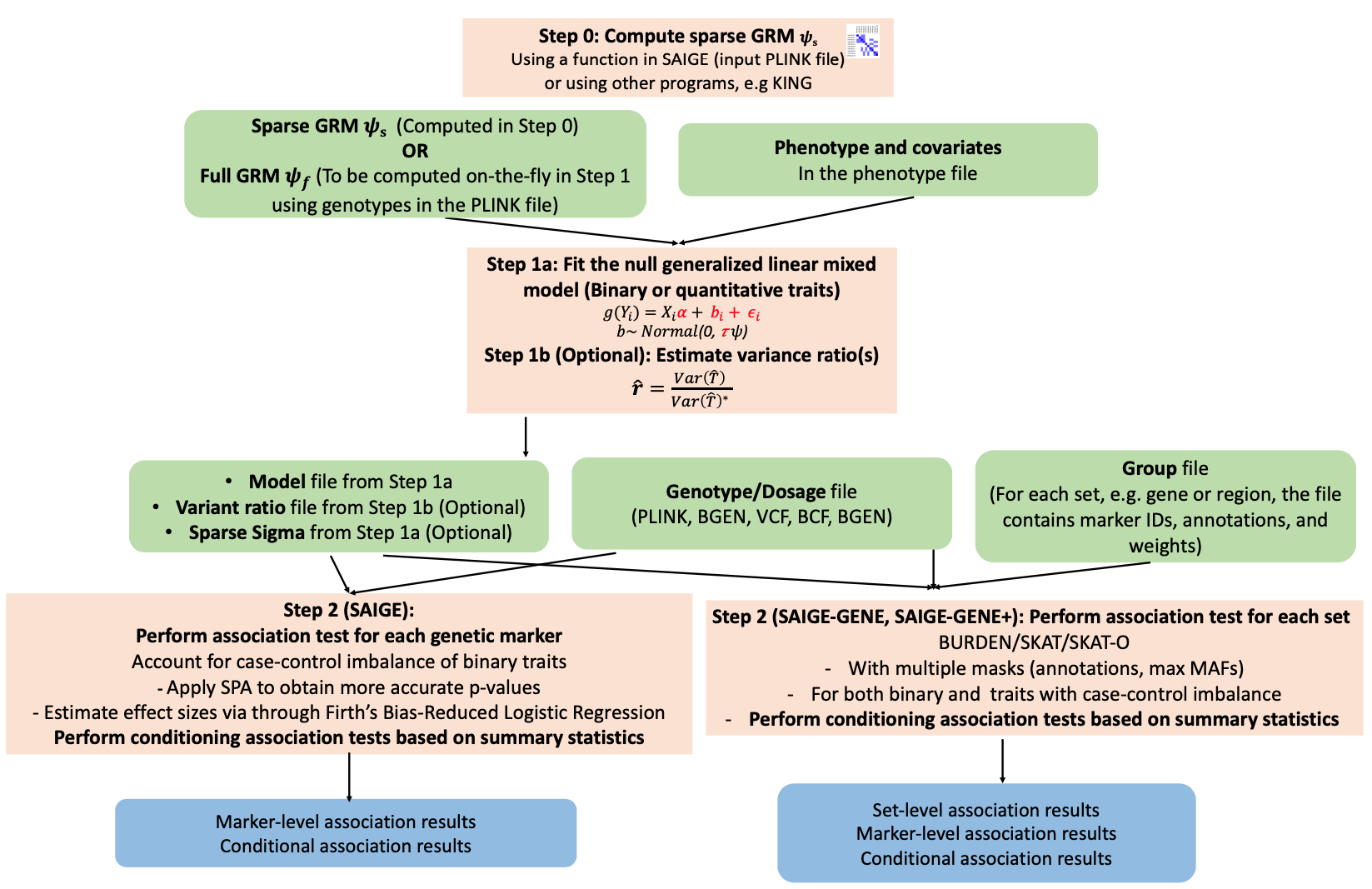
SAIGE 使用广义混合模型(GMM)来估计样本之间的相关性,并对每个基因位点进行全基因组关联分析。其核心思想是通过计算每对样本之间的遗传关系矩阵(GRM),从而调整样本间的相关性,避免这些关系对关联分析结果的干扰。
在分析过程中,SAIGE 会先使用 GRM 计算出样本间的相关性,并根据遗传相关性调整表型和基因型数据。接下来,使用 REML(限制最大似然法)或其他拟合方法进行模型估计,最后通过统计检验得到每个 SNP 与表型之间的关联。
2.1 计算遗传关系矩阵(GRM)
遗传关系矩阵(GRM)用于量化样本之间的基因型相似性。SAIGE 支持两种类型的 GRM:完整 GRM 和 稀疏 GRM。对于大规模数据集,稀疏 GRM 可以显著减少计算成本。我们可以使用 PLINK 等工具计算 GRM 文件,并将其作为输入提供给 SAIGE。
2.2 模型拟合与关联分析
SAIGE 使用 限制最大似然法(REML) 或其他拟合方法来估计基因型和表型之间的关系。对于稀有变异,SAIGE 还提供了 Firth 偏差减少逻辑回归,提高了小样本数据和稀有变异分析的准确性。